


1Dermatology Unit, 2Molecular Genetics Laboratory and 4Pathology Unit, Bambino Gesù Children’s Hospital, IRCCS, Piazza Sant’Onofrio, 4, IT-00165 Rome, and 3Molecular Medicine and Genetics AOU, Siena, Italy. E-mail: andrea.diociaiuti@opbg.net
#These authors contributed equally.
Accepted Mar 4, 2019; E-published Mar 5, 2019
X-linked ichthyosis (XLI, OMIM #308100) is the second most common form of inherited ichthyosis after ich-thyosis vulgaris. It almost exclusively affects males and the estimated prevalence ranges from 1 in 1,500 to 1 in 6,000 males (1–3). XLI is caused by mutations in the STS gene on chromosome Xp22.31 encoding for the steroid sulfatase (STS) enzyme (1, 4). Most patients (85–90%) carry a genomic deletion comprising the entire STS gene, while point mutations or partial deletions account for about 10% of cases. XLI manifests more frequently in the neonatal period with generalized whitish, lamellar desquamation, which over time is replaced by the typical polygonal, dark scales that are more prominent on legs and arms (1, 4).
We report an exceptional case of XLI in an adopted Indian girl homozygous for a previously undescribed nonsense mutation in the STS gene.
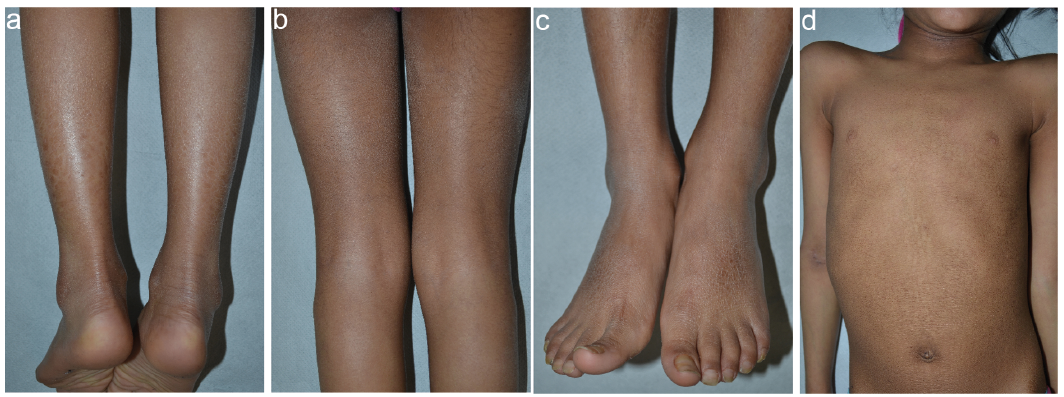
The patient is an 8-year-old female child of Indian origin who was referred to our rare skin disease center for suspicion of ichthyosis. She had been adopted 1 year before, and personal and family histories were unavailable. Physical examination showed diffuse dark scales which were larger on the legs and foot dorsa, smaller on the arms and thighs, and arranged in a reticular pattern on the thorax and abdomen (Fig. 1). The scalp showed fine, light desquamation, while the flexural creases presented minimal hyperkeratosis (Fig. 1b). The palmoplantar surfaces were unaffected. In addition, the child had mild signs of atopic dermatitis with lichenification on the right antecubital fossa, neck (Fig. 1d), perioral area and eyelids. She was in ophthalmological follow-up for reduced visual acuity due to retinopathy of prematurity. Neuropsychiatric evaluation did not reveal behavioural abnormalities. Her weight was below the 3rd percentile and her height between the 3rd and the 10th percentile. Laboratory examinations showed elevated total IgE (248 IU/ml) and vitamin D deficit (9.3 ng/dl), for which vitamin D supplementation was started.
Fig. 1 Clinical features of an 8-year-old Indian girl affected with X-linked ichthyosis. Large, polygonal, brownish scales on the calves (a); fine grayish scales on the back of thighs and minimal hyperkeratosis of the popliteal folds (b); hyperkeratosis and small to medium size scales on the foot dorsa and ankles (c); adherent, small scales arranged in a reticular pattern on the abdomen, lichenification on the antecubital right fossa and on the neck (d).
Following informed consent, a skin biopsy was obtained, together with blood sampling for genetic analysis. Histopathological examination showed compact hyperkeratosis, a normal granular layer, and follicular plugging. Immunohistochemistry revealed filaggrin expression similar to control skin. Ultrastructural examination did not show any lipid droplets, cholesterol clefts or membranous structures in the epidermal granular and horny layers, where numerous melanosome remnants were observed. Keratohyaline granules were normal in size and distribution.
Variant analysis was performed using a customized next generation sequencing (NGS) panel containing all known inherited ich-thyosis genes (5) except FLG. A homozygous nonsense mutation c.1116G>A (p.Trp372*) in exon 8 of the STS gene (NM_000351.4, NP_000342.2) (Fig. S1a) was identified. Sanger sequencing confirmed the presence of the homozygous G>A transversion (Fig. S1b). Mutation p.Trp372* has not been previously reported, and is not recorded in any database of human genetic variations (1,000 Genomes, Exome Variant Server and Exome Aggregation Consortium database – ExAC). Moreover, the dosage of STS activity in leukocyte homogenates using a previously described fluorimetric assay (6) was 0.47 nmol/17 h/mg (normal values in females: 2.06–3.77 nmol/17 h/mg), confirming STS deficiency. Thus, XLI was diagnosed. Subsequent MLPA analysis (P160-C1 STS MLPA kit, MRC-Holland, Amsterdam, the Netherlands) showed normal peak height (Fig. S1c), confirming the presence of both alleles at the STS locus.
Due to unavailability of patient’s parents, an SNP array (BeadChip 850K, Illumina, San Diego, CA, USA) was carried out to indirectly evaluate consanguinity and to exclude maternal X
chromosome isodisomy or other rearrangements. A homozygosity region of about 30 Mb was detected at Xp22.33p21.1. Multiple additional homozygosity blocks, accounting for about 25% of the autosome, were also identified.
XLI is almost always observed in males, while only a handful of cases have been described in females (7–12). In our adopted girl, the unavailability of personal and family histories, in particular the patient’s age at disease onset, its course, and the likely presence of an affected father, have hampered clinical diagnosis. On the other hand, sparing of palmoplantar surfaces is considered one of the most reliable clinical findings supporting a diagnosis of XLI (1, 4, 13). Clinical features were detailed in only 4 out of 8 previously reported cases of XLI in females (8, 10, 11). All these patients presented dark scales that were more prominent on extensor surfaces of extremities. Apart from one case that had increased skin markings of the soles (8), they all showed unaffected palmoplantar regions. Flexural areas were either normal (two cases) or slightly involved (two cases). Disease onset was at birth (two cases) or shortly after (two cases). With the exception of one severely affected woman who carried both STS and FLG mutations (10), the overall clinical features in females were similar to those in male XLI patients from the same family. Our patient also had a mild atopic dermatitis. Although we did not search for mutation(s) in FLG, the normal expression of filaggrin by immunohistochemistry is not in favour of FLG mutation(s).
In our case, the use of a NGS customized panel resulted in the rapid identification of a previously undescribed nonsense homozygous mutation, p.Trp372*, in the STS gene. The pathogenic role of the mutation was further confirmed by the detection of highly reduced STS activity in the patient’s leukocytes. To date, 25 different point mutations have been reported in patients affected by XLI (4). The p.Trp372* is a new mutation that changes the original sequence coding the amino acid tryptophan to a stop codon, breaking-down the regular polypeptide synthesis. Two additional mutations affecting the same tryptophan residue have been described: p.W372R and p.W372S. Our finding supports the hypothesis that, in addition to random changes, the STS gene may also be prone to recurrent defects in some particular regions. Moreover, more than 30% of known point mutations fall within exon 8.
XLI can be observed in patients affected with Turner syndrome. Indeed, the first female case of XLI was described in a Turner syndrome patient (45,X) in 1971 (7). In this instance, XLI diagnosis was made on the basis of the clinical presentation and family pedigree showing 7 affected males with an X-linked inheritance pattern. In our patient, the detection of two X chromosomes by MLPA and SNP array analysis allowed us to exclude Turner syndrome. Homozygosity for an X-linked mutation is the most frequent cause of XLI in women. In a consanguineous family with 3 affected sisters described in 1981, the diagnosis was based on the absence of STS activity (8). Subsequently, 4 female cases have been molecularly characterized: they were all due to homozygous STS genomic deletions (9–12). Three out of 4 families were consanguineous, and a founder effect was hypothesized in the 4th kindred. In our patient, the presence of multiple large homozygosity blocks on the X chromosome and autosomes is a robust indication of a high degree of consanguinity. In addition, SNP array findings exclude X chromosome maternal isodisomy, which could theoretically cause XLI in females. Altogether, our molecular findings strongly suggest that our patient inherited the same mutation from consanguineous parents, i.e. an affected hemizygous father and a heterozygous carrier mother. However, we cannot formally prove our hypothesis due to the lack of information on family history in our adopted female child.
In conclusion, our patient represents the first reported female case of XLI caused by a point mutation. Our findings also show the usefulness of NGS technology in diagnosing very rare and difficult ichthyosis cases, in particular in the absence of patient and family histories. Moreover, the consanguinity of the parents has been clarified by our SNP array, supporting the value of combining high-throughput molecular genetic technologies to improve genetic counselling.


 Click to show fullsize
Click to show fullsize